Remediation implications for medical device manufacturers in changing regulatory landscape
Blog: Capgemini CTO Blog
The life sciences sector is experiencing unprecedented regulatory change affecting organizations involved in pharmaceuticals, medical devices, and in-vitro diagnostics with the EU MDR. It is necessary for medical device companies to proactively prepare for these changes as the impact on their current and future business could be significant. The aim of the new regulation is to ensure that products are effective and safe throughout the product lifecycle. A manufacturer’s portfolio of products should be reviewed and assessed against the new regulations and future requirements.
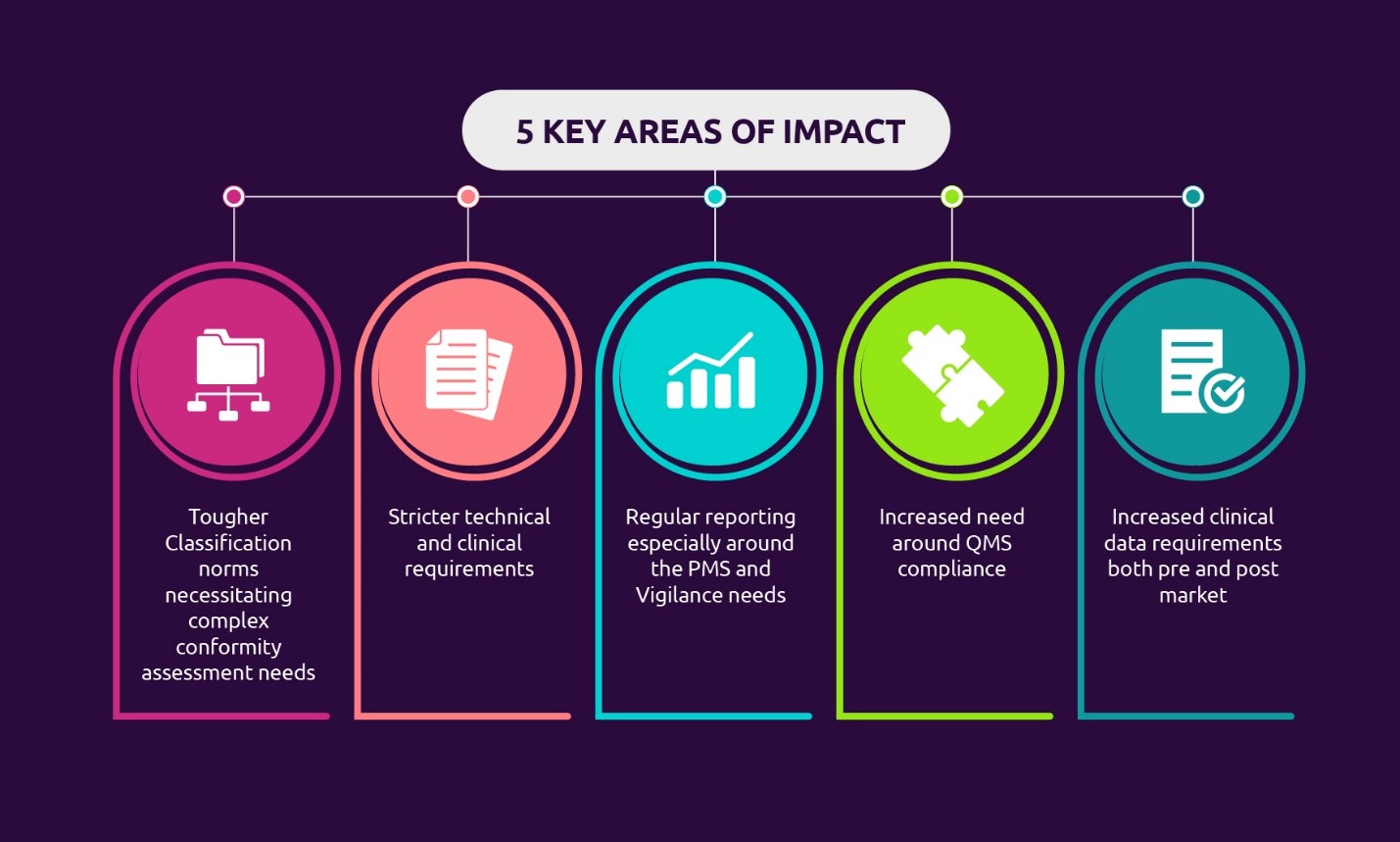
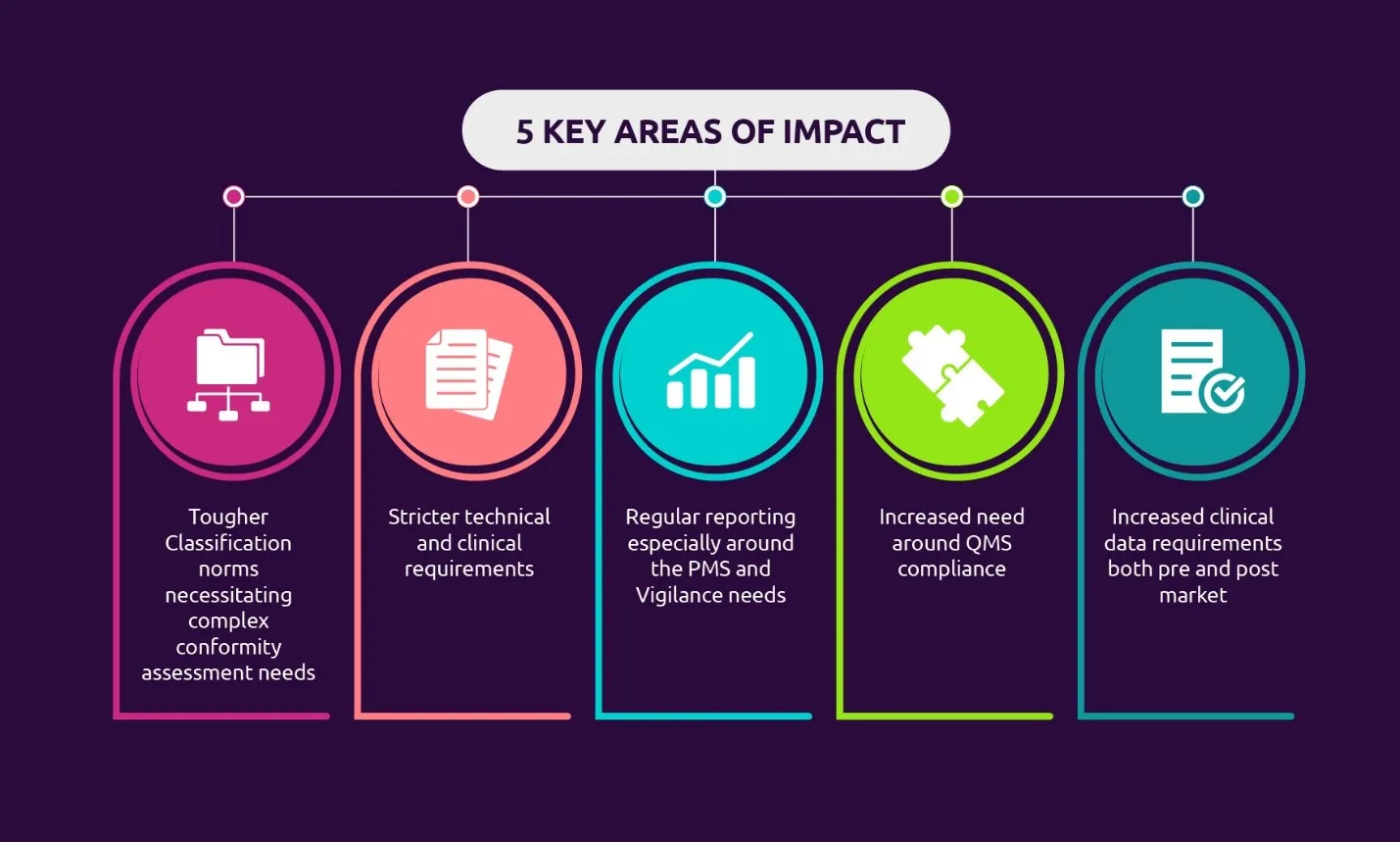
Tougher classification norms
With the new regulation, products that are classified as accessories can now be covered under the definition of a medical device. Besides, the MDR is bringing some products that were not classified as medical devices before MDR 2017/745 under the medical devices umbrella. This brings a major change to the QMS as compliance with the MDR is expected for the products that are classified as medical devices without any intended medical use. There may also be product lines for which the classification status will change and the oversight of notified bodies will be increased. Based on product classification changes, there will be significant implications for the technical documentation for the family of medical devices.
Stricter technical and clinical documentation
A new list of harmonized standards has been adopted for the MDR to ensure that the safety and performance of the medical devices match the latest technologies. The MDR has also brought in the common specifications for the devices that lack harmonized standards. This complete overhauling of standards affects the current compliance status of the products. The MDD’s essential requirements are now replaced with the MDR’s General Safety and Performance Requirements, which demands a revisit of the safety and performance characteristics of the medical devices. There has to be a gap assessment to address the new requirements added in the GSPR to adopt the latest harmonized standards. Thereby, a mandatory update in the design history file (DHF) and a technical file is needed. Additionally, a unique device identification will be implemented for differences between the FDA and EU classification if the products are to be marketed globally.
Regular reporting of PMS and vigilance requirements
With key focus being on the safety and effectiveness of devices over the entire lifecycle of the product, manufacturers are required to thoroughly monitor the safety profile of the products placed on the market through the implementation of a robust preapproval system, post-market surveillance (PMS), and vigilance systems with strict timelines to report adverse events. The manufacturers are expected to periodically collect the post-production data, evaluate and summarize the findings from the field, implement and maintain a risk management system throughout the lifecycle of a medical device. Moreover, the timeline for serious event reporting has been reduced and regular safety updates should be done for EUDAMED, which is a database where all data is periodically updated to provide the necessary market transparency, vigilance, post-market surveillance for the competent authorities, and public.
Increased need for QMS compliance and clinical data requirements
With significant mergers and acquisitions and other business reasons, companies have more than one QMS to deal with. For upgrading the QMS to the new regulatory environment, there is expected to be a significant effort. Given this, medical device companies are looking for partners to support them in rationalizing the QMS across their different organizations and upgrade them to the latest regulatory needs.
Medical device manufacturers need to ensure the process of clinical evaluation reports (CER) with periodic updates in terms of PMCF, PSUR, SSCP are robust enough to be updated annually as part of the post-market surveillance and vigilance processes.
As the change will impact the entire product lifecycle, organizations should be prepared for the increased need for clinical data requirements, with an emphasis on post-market surveillance and vigilance. Portfolio rationalization, understanding the new clinical evidence, gap analysis, and getting a perspective of the remediation needs will require an enterprise-wide approach. A multi-disciplinary and cross-functional team will be needed to take up and execute the requirements. The current systems and tools should be evaluated to address the new requirements and allowed for automation to enable aspects such as the EUDAMED database updates.
For over 18 years, Capgemini is working closely with leading global medical device companies across specialties as their extended R&D arm, with expertise in product and process remediation enabled by multi-disciplinary technical and domain skills. We are geared to address the compliance requirements for companies without impacting time to market for both existing and new products. Additionally, Capgemini brings in a structured approach to the entire process of remediation, from conducting gap analysis and arriving at a remediation plan to implementation and ongoing support around post-market requirements.
Visit our page to know more about remediation services for medical devices.
Author

Pradeep Kolankari – Sr. Director, Medical Devices and Lifesciences, Capgemini, Digital engineering and Manufacturing Services
Pradeep leads the solution team within the medical and healthcare practice having been associated with medical device industry in cardiology, neurology, imaging and IVD segments.